 Janki Darlami1
Janki Darlami1 Shweta Sharma2*1Mansarovar Global University, Department of Chemistry, Bhopal, Madhya Pradesh, India2Career College (Autonomous), Department of Chemistry, Bharat Heavy Electricals Limited, Bhopal, Madhya Pradesh, India
Shweta Sharma2*1Mansarovar Global University, Department of Chemistry, Bhopal, Madhya Pradesh, India2Career College (Autonomous), Department of Chemistry, Bharat Heavy Electricals Limited, Bhopal, Madhya Pradesh, IndiaQuantitative structure activity relationship (QSAR) is a widely used tool in rational drug design that establishes relationships between the physicochemical and topological descriptors of ligands and their biological activities. Obtained QSAR models help identify descriptors that play pivotal roles in the biological activity of ligands. This not only helps the prediction of new compounds with desirable biological activities but also helps with the design of new compounds with better activities and low toxicities. QSAR commonly uses lipophilicity (logP), hydrophobicity (logD), water solubility (logS), the acid–base dissociation constant (pKa), the dipole moment, the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), molecular weight (MW), molar volume (MV), molar refractivity (MR), and the kappa index as physicochemical parameters. Some commonly used topological indices in QSAR are the Wiener index, Platt index, Hosoya index, Zagreb indices, Balaban index, and E-state index. This review presents a brief description of the significance of the most extensively used physicochemical and topological parameters in drug design.
1 IntroductionA drug is defined as a chemical substance with a well-established and known structure that, when introduced, brings about a positive biological effect by restoring or modifying the physiology of the body (Benedetti, 2014). Developing a drug is a difficult, expensive, and tedious exercise. It takes approximately two decades and an average expense of US$1–2 billion to make a drug available at pharmacies. The high-risk nature of the process can be better comprehended by realizing that 90% of drug development projects fail once they enter the initial phase of a clinical trial. Possible causes reported for the failures are poor clinical efficacy (40%–50%), toxicity (30%), poor druggability (10%–15%), and strategic factors (10%). The biological activities of molecules primarily depend on their structure, physicochemical parameters, topological parameters, and the mechanism they take up. It is therefore important to have a better understanding of these parameters, which are significant in improving the activity of molecules, to avoid failures in drug design at the latter stages (Macalino et al., 2015).
In silico is an innovative computer-aided drug design approach that employs computational methods to identify potential drugs through the virtual screening of large chemical databases (Wang et al., 2015). The potential toxicity and druggability of candidate drugs are also tested in silico, which helps reduce the number of compounds that need to be evaluated in laboratories, resulting in a rational drug design and a 50% reduction in cost and time (Wang et al., 2015; Surabhi and Singh, 2018). The in silico approach has shown accelerated growth in recent decades because of the availability of large amounts of data and introduction of advanced computational methods supported by the latest AI techniques. Several drugs, such as darunavir, tipranavir, lopinavir, and ritonavir used to treat HIV/AIDS; oseltamivir and zanamivir to treat influenza; and imatinib, sorafenib, erlotinib, crizotinib, and ribociclib to treat cancer, were developed in silico. Rational drug design takes up two different courses: structure-based drug design and ligand-based drug design (LBDD) (Philip et al., 2007). LBDD is helpful when the experimental data and three-dimensional structures of target proteins are not available. It adopts the indirect line and studies the structural and physicochemical properties of known ligands that are pivotal for their biological activities (Acharya et al., 2011; Wilson and Lill, 2011). Quantitative structure activity relationship (QSAR) and pharmacophore are two widely used tools in LBDD.
2 Quantitative structure activity relationshipQSAR relates the chemical structures of ligands to their biological activities. Two- and three-dimensional structures of known ligands are evaluated to identify the molecular descriptors that are vital for the biological activity. Based on the similar descriptors, new ligands with similar structures are predicted to have better inhibitory activity and lower toxicity (Rudrapal and Egbuna, 2022).
3 Role of physicochemical descriptors in drug designDescriptors are the numeric expressions of the chemical characteristics of compounds (Rudrapal and Egbuna, 2022). Physicochemical descriptors of ligands that correlate well with the binding energy are significant for biological activity. Interaction of the ligand with the receptor and its energetic and entropic terms decide its biological activity. For example, the hydrophobicity of a compound indicates its dispersive and electrostatic interaction with the receptor, and as there is a close relationship between polarizability and the dispersive force and density of a charge on an atom and electrostatic forces, these factors have to be accounted for when calculating hydrophobicity (Ghose and Crippen, 1987). Some of the most common physicochemical properties are described in the following sections.
3.1 Octanal–water partition coefficient (lipophilicity logP and hydrophobicity logD)Lipophilicity is one of the main influencing factors in the transportation of a drug through a biological membrane in lipophilic medium (Raevsky, 2004; Davis and Leeson, 2023). The conventional method for determining lipophilicity is the partitioning of a drug between n-octanal and water (Raevsky, 2004), as follows:
LogP=log10Drugn−octanal /Drugwater.Most of the reported QSAR/Quantitative Structure Property Relationship (QSPR) models have an integrated logP term that indicates the significance of lipophilicity in predicting the biological activity of a drug (Raevsky, 2004). The use of a 1-octanal–water pair to measure logP was first reported, and Hansch et al. developed a hydrophobic substituent constant π to determine it (Fujita et al., 1964). Different approaches to determining the value of logP have been reported, such as the fragmental approach, which was found to be an easier method for determining logP (Leo et al., 1975; Rekker, 1977), and Computer Automated Structure Evaluation of Molecules (MCASE) group contribution and atomic contribution approaches, which found the logP values of the 935 and 893 compounds, respectively (Ghose and Crippen, 1987; Klopman and Wang, 1991). Other methods reported to model logP are molecular fingerprints (Liu and Zhou, 2008), multiple linear regression, support vector machines, radial neural networks and artificial neural networks (Chen, 2009), and E-state indices (Kier and Hall, 1999). Hydrophobicity is expressed as a distribution constant through the partition of a drug at a certain pH value (Dearden, 2012; Davis and Leeson, 2023), as follows:
Log D7.4=logP–log1+10pH – pKa (in acidic medium).
Log D7.4=logP –log1+10pKa − pH (in basic medium).
Log D=logP–log1+10pKa1−pH+10pKa2−pH (for a zwitterion) (Davis and Leeson, 2023).
3.2 Water solubility (logS)Several studies have been performed to determine the value of water solubility from the molecular and physicochemical properties of drugs to evaluate their Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) profile (Raevsky, 2004; Wenlock and Barton, 2013). Water solubility and hydrophobicity are inversely related to each other and can be estimated through the relationship given as (Hansch et al., 1968; Yalkowsky and Valvani, 1980)
Log S=−1.07logP+0.67.With an increase in the hydrogen bonds effect, the solubility in water increases and decreases with the increase in steric bulk. Applying linear and nonlinear relationships, different physicochemical descriptors were used to determine the value of logS. These descriptors include molecular volume, molecular weight, polar surface area, fractional hydrogen donor surface area, molecular polarizability, molecular refractivity, Linear Free-Energy Relationships (LSER) descriptors, log P, dipole moment, heat of formation, total energy, electronic energy, the number of hydrophilic rotatable bonds, the number of H-B donor–acceptors, and E-state (Raevsky, 2004). As most of the drugs are in solid form, crystal lattice energy is calculated, which is related to its melting point (Dearden, 2012). Therefore, aqueous solubility can be modeled using logP and the melting point (Yalkowsky and Valvani, 1980) as follows:
Log S=0.87–1.05logP–0.012MP.3.3 The acid–base dissociation constant (pKa)The acid–base dissociation constant is another key factor that affects the ADMET profile of a drug by influencing its lipophilicity, aqueous solubility, and permeability. As drugs are either a weak acid or base, the calculation of their dissociation constant assists in understanding their ionic forms across different pH values (Manallack, 2007). The dissociation constant is calculated using the following formula (Wenlock and Barton, 2013):
pKa=−log10Ka≈−log10H+aq BaseaqAcidaq.Generally, the pKa, Hammett substituent constant (σ), and acid dissociation constant are related to each other as (Livingstone, 2003; Dearden, 2012)
pKaderivative=pKaparent–ρσ (where ρ = series constant).
4 Role of topological descriptors in drug designTopological indices or descriptors are the result of the successful application of mathematical “graph theory” in chemistry, in which chemical structures are represented as graphs. These two-dimensional graphs are then used to obtain numeric values for topological indices (Gozalbes et al., 2002). The calculation of topological indices is easy and fast; they correlate quite well with the physicochemical properties and biological activities of compounds and are therefore widely used in QSAR/QSPR (Dearden, 2017). The size and shape of the molecule, number of rings present, branching of the carbon chain, π-electron energy, molar refractivity, molar volume, heat of formation, and heat of vaporization are molecular properties that decide the significant interaction between the ligand and receptor. Some of the most common topological indices are described in the following sections.
4.1 Wiener index (W) and Platt indexThe Wiener index refers to the C-C bonds between all pairs of C-atoms in alkane. This index has been used to determine the boiling points of some straight-chain and branched-chain alkanes (Dearden, 2017). Additionally, the Wiener index has been used to predict the toxicity of some nitrobenzenes to T. pyriformis (Ivanciuc, 2000). The Platt index is the total sum of adjacent bonds for each atom in alkane. This index has been used in several QSAR models to study the anti-leishmanial activity of phloroglucinol–terpene adducts (Bharate and Singh, 2011).
4.2 Hosoya index (Z)This index refers to the total number of non-adjacent bonds in a compound (Dearden, 2017). The utility of the Hosoya index in combination with other descriptors was reported in a QSAR study to predict the anti-Alzheimer activity of a set of N-aryl derivatives (Solomon et al., 2009).
4.3 Zagreb indicesThe number of non-H-bonds formed by heavy atoms in a compound is squared, and the total sum of these squares is calculated as the Zagreb index (Dearden, 2017). The Zagreb group parameters M1 and M2 were found to be useful in predicting the pharmacokinetic parameters of cephalosporins in humans (Dureja et al., 2008).
4.4 Balaban index (J)The Balaban index is a connectivity index that takes the sum of average distances between atoms in a molecule into account (Dearden, 2017). The index significantly modeled the QSAR study of benzenesulfonamide as a carbonic anhydrase inhibitor (Thakur et al., 2004).
4.5 E-state indexThis index was developed by Kier and Hall (1999) by integrating the electronic and topological features that characterize the atomic level of interaction inside the molecule (Gozalbes et al., 2002; Dearden, 2017). Successful QSAR modeling of benzofuran derivatives (melatonin receptor) and hydroxyphenylureas (antioxidants) has been reported using a combination of physicochemical descriptors with the E-state index (Sengupta et al., 2004; Ray et al., 2010). Selecting suitable physicochemical and topological parameters of compounds is crucial in the identification of the inhibitory activities of compounds (depicted in Supplementary Table S1).
5 ConclusionOwing to the cost-effective and time-efficient nature of rational drug design, almost every drug development project has adopted the in silico method, which uses several computational and statistical techniques that are constantly being improved. Therefore, the success of these projects requires deep knowledge of and skill in using these techniques efficiently. Moreover, advancements in genome sequencing and crystallography have resulted in an increasing amount of data becoming available in databanks. While performing QSAR, it is essential to identify and select suitable parameters, as this determines the credibility and reliability of the model. The selection of an excessive number of descriptors in QSAR/QSPR should be avoided as it can result in overfitting. In this review, we have provided an overview of the significance of selecting and using suitable physicochemical and topological descriptors in predicting the biological activities of compounds. The interrelated role of physicochemical and topological descriptors is depicted in Figure 1.
Figure 1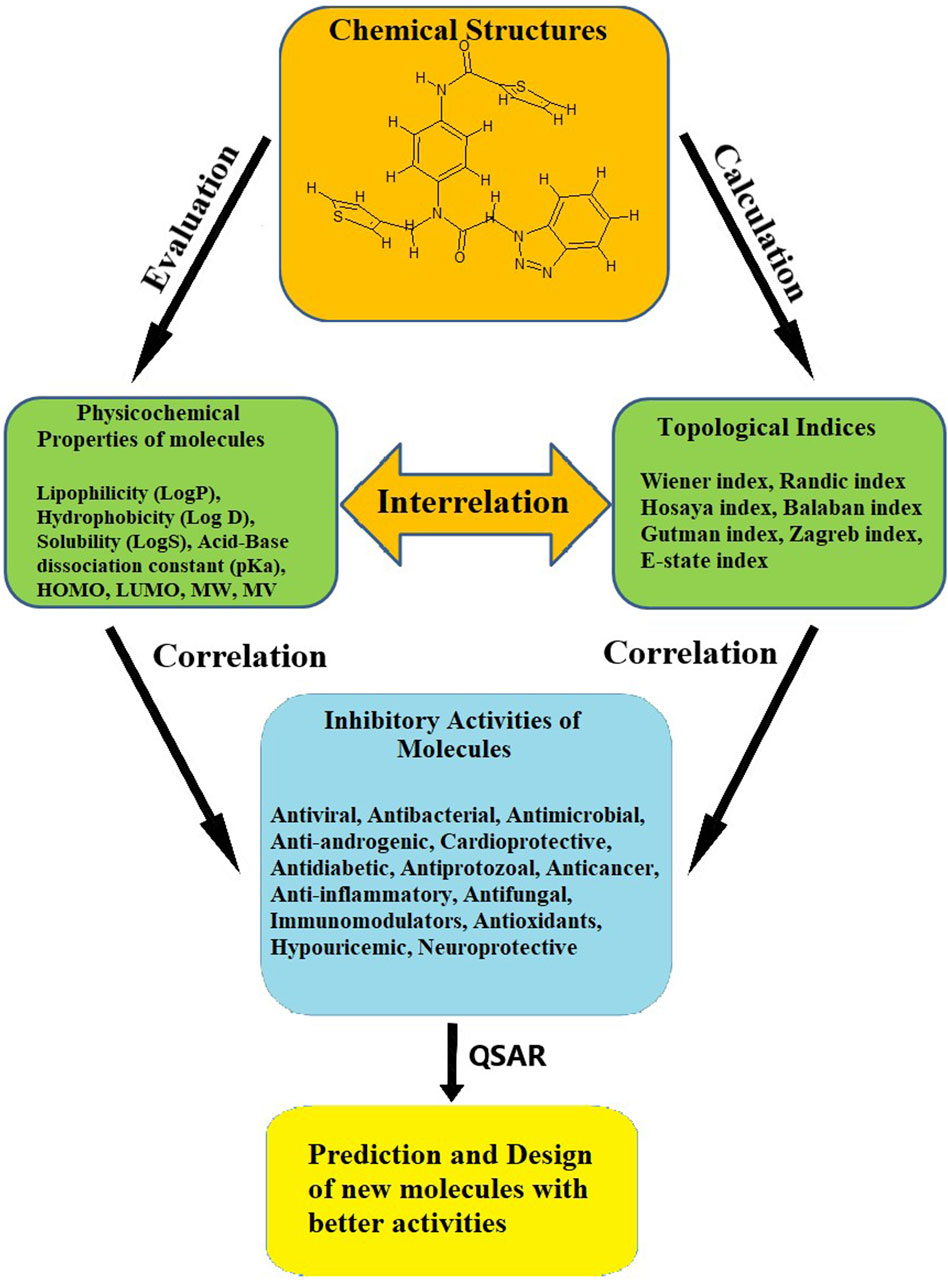
Figure 1. Interrelated significance of physicochemical properties and the topological index.
QSAR, machine learning, and virtual screening (VS) are artificial intelligence (AI) approaches that have been successfully used in rational drug design to reduce the time and cost. Recently, deep learning (DL), a new AI approach, has gained popularity due to its efficiency in analyzing big data. DL-de novo and DL-VS approaches are widely used. The prediction of the three-dimensional structure of proteins using one-dimensional amino acid sequences, drug DSP-1181 entering clinical trials, and the prediction of anti-Plasmodium falciparum are a few successes of DL. Owing to its efficiency in dealing with massive data, it is expected that DL will be useful in drug repurposing, genome mining, and toxicity prediction.
Author contributionsJD: Writing–review and editing. SS: Conceptualization, formal analysis, supervision, and writing–original draft.
FundingThe authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2024.1424402/full#supplementary-material
ReferencesAcharya, C., Coop, A., Polli, J. E., and Mackerell, A. D. (2011). Recent advances in ligand-based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Curr. computer-aided drug Des. 7 (1), 10–22. doi:10.2174/157340911793743547
PubMed Abstract | CrossRef Full Text | Google Scholar
Benedetti, F. (2014). Drugs and placebos: what’s the difference? EMBO Rep. 15 (4), 329–332. doi:10.1002/embr.201338399
PubMed Abstract | CrossRef Full Text | Google Scholar
Bharate, S., and Singh, I. P. (2011). Quantitative structure-activity relationship study of phloroglucinol-terpene adducts as anti-leishmanial agents. Bioorg. Med. Chem. Lett. 21, 4310–4315. doi:10.1016/j.bmcl.2011.05.053
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, H.-F. (2009). In silico log P prediction for a large data set with support vector machines, radial basis neural networks and multiple linear regression. Chem. Biol. drug Des. 74 (2), 142–147. doi:10.1111/j.1747-0285.2009.00840.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Davis, A. M., and Leeson, P. D. (2023). “Physicochemical properties,” in The handbook of medicinal chemistry. Editors S. E. Ward, and A. Davis (Cambridge, United Kingdom: The Royal Society of Chemistry), 0. doi:10.1039/9781788018982-00001
CrossRef Full Text | Google Scholar
Dearden, J. C. (2012). “Prediction of physicochemical properties,” in Computational toxicology: volume I. Editors B. Reisfeld, and A. N. Mayeno (Totowa, NJ: Humana Press), 93–138. doi:10.1007/978-1-62703-050-2_6
CrossRef Full Text | Google Scholar
Dearden, J. C. (2017). “The use of topological indices in QSAR and QSPR modeling,” in Advances in QSAR modeling: applications in pharmaceutical, chemical, food, agricultural and environmental sciences. Editor K. Roy (Cham: Springer International Publishing), 57–88. doi:10.1007/978-3-319-56850-8_2
CrossRef Full Text | Google Scholar
Dureja, H., Gupta, S., and Madan, A. K. (2008). Topological models for prediction of pharmacokinetic parameters of cephalosporins using random forest, decision tree and moving average analysis. Sci. Pharm. 76, 377–394. doi:10.3797/scipharm.0803-30
CrossRef Full Text | Google Scholar
Fujita, T., Iwasa, J., and Hansch, C. (1964). A new substituent constant, π, derived from partition coefficients. J. Am. Chem. Soc. 86 (23), 5175–5180. doi:10.1021/ja01077a028
CrossRef Full Text | Google Scholar
Ghose, A. K., and Crippen, G. M. (1987). Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 27 (1), 21–35. doi:10.1021/ci00053a005
PubMed Abstract | CrossRef Full Text | Google Scholar
Gozalbes, R., Doucet, J. P., and Derouin, F. (2002). Application of topological descriptors in QSAR and drug design: history and new trends. Curr. Drug Targets - Infect. Disord. 2 (1), 93–102. doi:10.2174/1568005024605909
PubMed Abstract | CrossRef Full Text | Google Scholar
Hansch, C., Quinlan, J. E., and Lawrence, G. L. (1968). Linear free-energy relationship between partition coefficients and the aqueous solubility of organic liquids. J. Org. Chem. 33, 347–350. doi:10.1021/jo01265a071
CrossRef Full Text | Google Scholar
Ivanciuc, O. (2000). QSAR comparative study of wiener descriptors for weighted molecular graphs. J. Chem. Inf. Comput. Sci. 40 (6), 1412–1422. doi:10.1021/ci000068y
PubMed Abstract | CrossRef Full Text | Google Scholar
Kier, L. B., and Hall, L. H. (1999). Molecular structure description: the electrotopological state. Available at: https://api.semanticscholar.org/CorpusID:92930114.
Google Scholar
Kirmani, S. A. K., Ali, P., and Azam, F. (2021). Topological indices and QSPR/QSAR analysis of some antiviral drugs being investigated for the treatment of COVID-19 patients. Int. J. Quantum Chem. 121 (9), e26594. doi:10.1002/qua.26594
PubMed Abstract | CrossRef Full Text | Google Scholar
Klopman, G., and Wang, S. (1991). A computer automated structure evaluation (CASE) approach to calculation of partition coefficient. J. Comput. Chem. 12 (8), 1025–1032. doi:10.1002/jcc.540120815
CrossRef Full Text | Google Scholar
Leo, A., Jow, P. Y., Silipo, C., and Hansch, C. (1975). Calculation of hydrophobic constant (log P) from pi and f constants. J. Med. Chem. 18 (9), 865–868. doi:10.1021/jm00243a001
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, R., and Zhou, D. (2008). Using molecular fingerprint as descriptors in the QSPR study of lipophilicity. J. Chem. Inf. Model. 48 (3), 542–549. doi:10.1021/ci700372s
PubMed Abstract | CrossRef Full Text | Google Scholar
Livingstone, D. J. (2003). Theoretical property predictions. Curr. Top. Med. Chem. 3 (10), 1171–1192. doi:10.2174/1568026033452078
PubMed Abstract | CrossRef Full Text | Google Scholar
Macalino, S. J. Y., Gosu, V., Hong, S., and Choi, S. (2015). Role of computer-aided drug design in modern drug discovery. Archives Pharmacal Res. 38 (9), 1686–1701. doi:10.1007/s12272-015-0640-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Manallack, D. T. (2007). The pK(a) distribution of drugs: application to drug discovery. Perspect. Med. Chem. 1, 1177391X0700100. doi:10.1177/1177391x0700100003
CrossRef Full Text | Google Scholar
Narasimhan, B., Judge, V., Narang, R., Ohlan, R., and Ohlan, S. (2007). Quantitative structure–activity relationship studies for prediction of antimicrobial activity of synthesized 2,4-hexadienoic acid derivatives. Bioorg. Med. Chem. Lett. 17 (21), 5836–5845. doi:10.1016/j.bmcl.2007.08.037
PubMed Abstract | CrossRef Full Text | Google Scholar
Philip, P., Dixit, A., and Saxena, A. K. (2007). Computer-aided drug design: integration of structure-based and ligand-based approaches in drug design. Curr. Comput. - Aided Drug Des. 3 (2), 133–148. doi:10.2174/157340907780809516
CrossRef Full Text | Google Scholar
Raevsky, O. A. (2004). Physicochemical descriptors in property-based drug design. Mini Rev. Med. Chem. 4 (10), 1041–1052. doi:10.2174/1389557043402964
PubMed Abstract | CrossRef Full Text | Google Scholar
Rasheed, M. W., Mahboob, A., and Hanif, I. (2023). An estimation of physicochemical properties of heart attack treatment medicines by using molecular descriptor’s. South Afr. J. Chem. Eng. 45, 20–29. doi:10.1016/j.sajce.2023.04.003
CrossRef Full Text | Google Scholar
Ray, S., Roy, P. P., Sengupta, C., and Roy, K. (2010). Exploring QSAR of hydroxyphenylureas as antioxidants using physicochemical and electrotopological state atom parameters. Mol. Simul. 36 (6), 484–492. doi:10.1080/08927021003664058
CrossRef Full Text | Google Scholar
Rekker, R. F. (1977). The hydrophobic fragmental constant, its derivation and application: a means of characterizing membrane systems. Available at: https://api.semanticscholar.org/CorpusID:94065099.
Google Scholar
Rudrapal, M., and Egbuna, C. (2022) Computer aided drug design (CADD): from ligand-based methods to structure-based approaches. Elsevier.
Google Scholar
Sengupta, C., Leonard, J. T., and Roy, K. (2004). Exploring QSAR of melatonin receptor ligand benzofuran derivatives using E-state index. Bioorg. \and Med. Chem. Lett. 14 (13), 3435–3439. doi:10.1016/j.bmcl.2004.04.073
PubMed Abstract | CrossRef Full Text | Google Scholar
Solomon, K. A., Sundararajan, S., and Abirami, V. (2009). QSAR studies on N-aryl derivative activity towards alzheimer’s disease. Molecules 14, 1448–1455. doi:10.3390/molecules14041448
PubMed Abstract | CrossRef Full Text | Google Scholar
Surabhi, S., and Singh, B. K. (2018). Computer aided drug design: an overview. J. Drug Deliv. Ther. 8 (5), 504–509. doi:10.22270/jddt.v8i5.1894
CrossRef Full Text | Google Scholar
Thakur, A., Thakur, M., Khadikar, P. V., Supuran, C. T., and Sudele, P. (2004). QSAR study on benzenesulphonamide carbonic anhydrase inhibitors: topological approach using Balaban index. Bioorg. Med. Chem. 12 (4), 789–793. doi:10.1016/j.bmc.2003.10.058
PubMed Abstract | CrossRef Full Text | Google Scholar
Wang, Y., Xing, J., Xu, Y., Zhou, N., Peng, J., Xiong, Z., et al. (2015). In silico ADME/T modelling for rational drug design. Q. Rev. Biophysics 48 (4), 488–515. doi:10.1017/S0033583515000190
PubMed Abstract | CrossRef Full Text | Google Scholar
Wardecki, D., Dołowy, M., and Bober-Majnusz, K. (2023). Evaluation of the usefulness of topological indices for predicting selected physicochemical properties of bioactive substances with anti-androgenic and hypouricemic activity. Molecules 28, 5822. doi:10.3390/molecules28155822
PubMed Abstract | CrossRef Full Text | Google Scholar
Wenlock, M. C., and Barton, P. (2013). In silico physicochemical parameter predictions. Mol. Pharm. 10 (4), 1224–1235. doi:10.1021/mp300537k
PubMed Abstract | CrossRef Full Text | Google Scholar
Wilson, G. L., and Lill, M. A. (2011). Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 3 (6), 735–750. doi:10.4155/fmc.11.18
PubMed Abstract | CrossRef Full Text | Google Scholar
Yalkowsky, S. H., and Valvani, S. C. (1980). Solubility and partitioning I: solubility of nonelectrolytes in water. J. Pharm. Sci. 69 (8), 912–922. doi:10.1002/jps.2600690814
PubMed Abstract | CrossRef Full Text | Google Scholar
Keywords: ligand-based drug design, QSAR, QSPR, physicochemical descriptor, topological descriptor
Citation: Darlami J and Sharma S (2024) The role of physicochemical and topological parameters in drug design. Front. Drug Discov. 4:1424402. doi: 10.3389/fddsv.2024.1424402
Received: 28 April 2024; Accepted: 31 May 2024;Published: 09 July 2024.
Edited by:
Rajesh Kumar Pathak, Chung-Ang University, Republic of KoreaReviewed by:
Neetesh Pandey, Columbia University, United StatesSutanu Nandi, University of Colorado Anschutz Medical Campus, United StatesVaishali Patil, Chaudhary Charan Singh University, IndiaCopyright © 2024 Darlami and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shweta Sharma, shwetasharma2703@gmail.com