来源:雪球App,作者: 六日行研社,(https://xueqiu.com/4812095430/299104395)


药苑杂谈YAOYUANZATAN
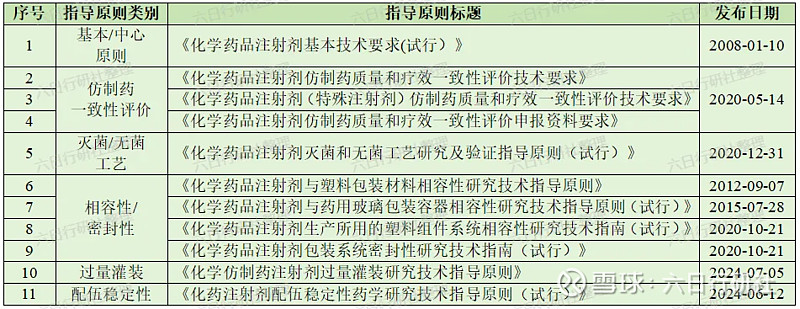
2017年12月22日CDE发布《已上市化学仿制药(注射剂)一致性评价技术要求(征求意见稿)》,标志着继口服固体制剂之后,注射剂一致性评价工作序幕拉开。关于化药仿制注射剂开发与一致性评价的技术要求,CDE发布了多篇指导原则供申请人参考,小编按类别列出了以下主要相关指导原则,并在本篇文章中和大家一起学习其中的主要内容。
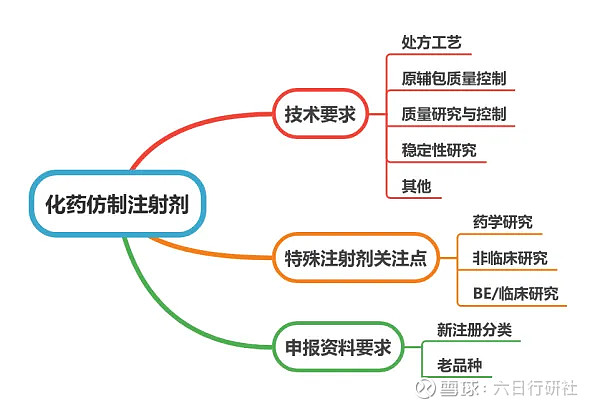
技术要求
小编以《化学药品注射剂仿制药质量和疗效一致性评价技术要求》的内容为主要框架,结合其他相关指导原则,将注射剂一致性评价技术要求分为:处方工艺、原辅包质量控制、质量研究与控制、稳定性研究、其他五个模块总结如下。
一、处方工艺
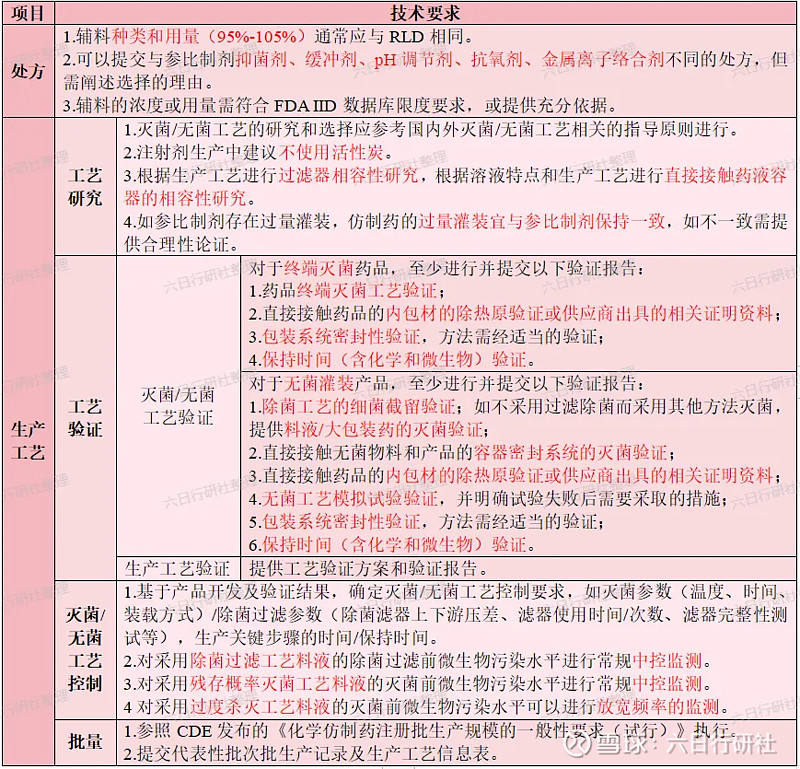
如上表提及的,关于注射剂灭菌/无菌工艺的研究和选择,《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》的要求如下:
灭菌工艺的选择一般按照灭菌工艺选择的决策树(详见下图)进行。
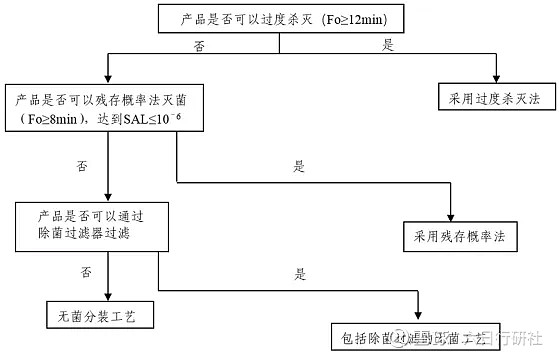
图片来源:《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》
湿热灭菌工艺是决策树中首先考虑的灭菌方法。注射剂的湿热灭菌工艺应首选过度杀灭法,即F0(标准灭菌时间)值大于等于12分钟的灭菌工艺;对热不稳定的药物,可以选择残存概率法,即F0值大于等于8分钟的灭菌工艺。两种湿热灭菌工艺都可以在实际生产中使用,具体选择哪种灭菌工艺,主要取决于产品的热稳定性。
注射剂应首选终端灭菌工艺。一般而言,需要通过各个方面的研究(研究建议如下表),使药物尽可能地可以采用湿热灭菌工艺。只有在理论和实践均证明即使采用了各种可行的技术方法之后,药物活性成分依然无法耐受湿热灭菌工艺时,才能选择无菌生产工艺。任何商业上的考虑均不能作为不采用具有最高无菌保证水平的最终灭菌工艺的理由。
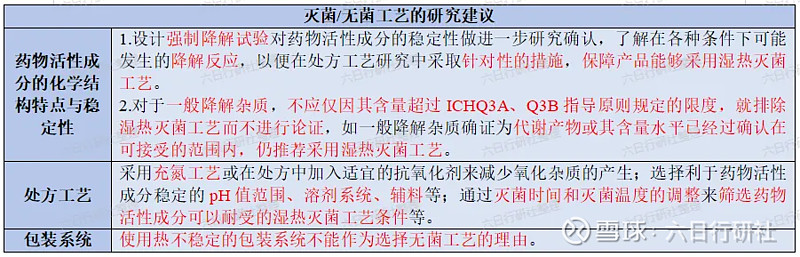
如经充分的研究(包括处方工艺研究、质量控制研究等)证实产品无法耐受终端灭菌工艺,则可考虑能否通过除菌过滤工艺进行过滤除菌,如果可以,可采用包含除菌过滤的无菌工艺;如果不可以,则可考虑采用无菌分装的全无菌工艺。
仿制注射剂选择的灭菌/除菌工艺,应能保证其无菌保证水平不低于参比制剂。如果参比制剂采用了无菌生产工艺,若仿制注射剂对其处方组成合理性、灭菌工艺产生的降解杂质等风险因素进行了全面的论证之后,也应按照灭菌工艺选择的决策树进行灭菌工艺的选择。
二、原辅包质量控制

三、质量研究与控制
1.根据目标产品的质量概况(QTPP)确立制剂的关键质量属性(CQA)。
2.有关物质:按照相关指导原则及国内外药典收载情况选择有关物质检查方法,并进行方法学验证。重点对制剂的降解产物进行研究,包括原料药的降解产物或者原料药与辅料和/或内包材的反应产物。原料药的工艺杂质一般不需要在制剂中进行监测或说明。
3.异构体:对于存在几何异构体和手性异构体等情况,根据产品特点和生产工艺等方面的研究,确定是否订入标准。
4.致突变杂质:判断是否可能产生潜在的致突变杂质,必要时进行针对性研究和控制。
5.元素杂质:根据ICH Q3D规定确定制剂中元素杂质的控制策略,包括原辅包、生产设备等可能引入的元素杂质。
6.申请产品应与参比制剂(原则上提供多批)进行全面的质量对比(含杂质谱对比),考察与一致性评价紧密相关的关键质量属性。
四、稳定性研究
1.注射剂稳定性研究内容包括影响因素试验、加速试验和长期试验,必要时进行中间条件试验。对低温不稳定建议进行低温试验和冻融试验。一般应提供不少于6个月的稳定性研究数据。
2.依据参比制剂说明书进行临床配伍稳定性研究,参比制剂说明书中配伍相关信息不明确的,建议参照《化药注射剂配伍稳定性药学研究技术指导原则(试行)》(相关内容总结如下表)新药要求开展配伍稳定性研究,并与参比制剂进行对比研究。
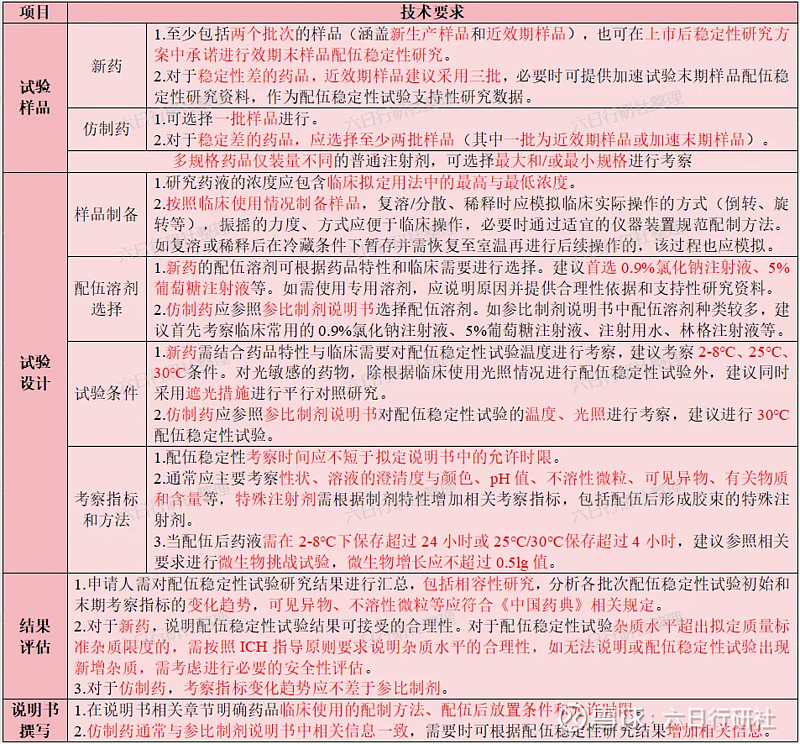
3.若注射剂处方中含有抗氧剂、抑菌剂等辅料,在稳定性研究中还要考察这些辅料含量的变化情况。
4.稳定性考察初期和末期进行无菌检查,其他时间点可采用包装系统密封性替代。包装系统密封性可采用物理完整测试方法(例如压力/真空衰减等)进行检测,并进行方法学验证。
5.仿制药的稳定性应不低于参比制剂。根据稳定性研究结果,参照参比制剂确定贮藏条件。
五、其他关注点
1.注射剂改规格:注射剂改规格指与参比制剂不同规格(包括原料药浓度不同)的注射剂,需充分论证改规格的科学性、合理性和必要性。
改规格的注射剂应与参比制剂适应症相同,并在参比制剂说明书规定的用量范围内使用,不得改变用法用量或适用人群。其规格一般不得小于单次最小给药剂量,也不得大于单次最大给药剂量。
2.说明书拟定:参考最新版参比制剂说明书合理拟定。
3.药品标准:收载检验项目应不少于中国药典规定,质量指标应不低于中国药典要求,若与中国药典不一致的,应提供合理充分的依据。
4.无需开展一致性评价的品种:氯化钠注射液、葡萄糖注射液、葡萄糖氯化钠注射液、注射用水、部分放射性药物等品种无需开展一致性评价,需进行质量提升研究,灭菌工艺、滤器与包材选择(含相容性研究)等应符合相关技术要求。
特殊注射剂关注点
特殊注射剂是指与普通注射剂相比,特殊注射剂的质量及其活性成分的体内行为受处方和工艺的影响较大,可能进一步影响制剂在体内的安全性和有效性,例如脂质体、静脉乳、微球、混悬型注射剂、油溶液、胶束等。
与普通注射剂仅需进行药学研究相比,特殊注射剂采用逐步递进的对比研究策略,首先在达到药学和非临床一致性基础上,还要进行生物等效性研究,必要时进行临床等效性研究。
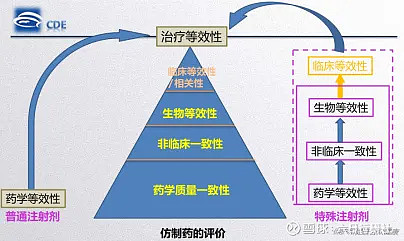
图片来源:CDE“特殊注射剂一致性评价技术要求”培训PPT
根据CDE发布的《化学药品注射剂(特殊注射剂)仿制药质量和疗效一致性评价技术要求》,特殊注射剂一致性评价的药学研究、非临床研究、BE/临床研究的关注点总结如下。
一、药学研究
1.除符合《化学药品注射剂仿制药质量和疗效一致性评价技术要求》外,还需参照FDA、EMA发布的特殊制剂相关技术要求,科学设计试验。对于FDA或EMA已公布指导原则的特定注射剂品种,建议参照其技术要求开展与参比制剂的对比研究。原则上应提供至少3批次参比制剂样品的质量对比考察数据。
2.处方原则上应与参比制剂一致,建议对辅料的型号及可能影响注射剂体内行为的辅料的CQA进行研究。
3.考察的关键质量属性可能包括但不限于:理化性质(如性状,黏度,渗透压摩尔浓度,pH值/酸碱度等),Zeta电位,粒子形态,粒径及分布,体外溶出/释放行为,游离和结合药物,药物晶型和结晶形态。
二、非临床研究
特殊注射剂进入体内后通常存在释药过程和体液成分吸附等因素,受试制剂与参比制剂处方和工艺的差异可能导致药物体内药代动力学行为发生改变,从而带来有效性和毒性的变化的,在开展人体生物等效性研究或临床试验前,应选择合适的动物种属进行非临床药代动力学对比研究,必要时进行组织分布比较。
三、BE/临床研究
1.对于静脉注射给药的普通注射剂,药学一致即可,不需要进行BE试验。
2.一般情况下,皮下、肌肉等非血管注射给药的注射剂需要进行BE研究,对于静脉注射给药的特殊注射剂需要进行BE研究,技术方法需具体分析。除CDE已发布的特殊注射剂BE研究指导原则外,对于FDA或EMA已公布BE指导原则的特定品种,建议参照研究。
3.对于特殊注射剂是否需要进一步展开随机对照临床试验,应基于药物特点,以及前期药学、非临床、人体生物等效性研究结果等讨论确定。对于人体生物等效性研究结果显示受试制剂与参比制剂不等效的,申请人应对受试制剂处方工艺进一步优化,重新开展对比研究。对于以下情况(不限于),建议开展随机对照临床试验研究,证明受试制剂与参比制剂的等效性:
(1)体循环中的药物浓度与疗效或安全性相关性较差,人体生物等效性研究不足以评价受试制剂与参比制剂的疗效、安全性一致。
(2)缺乏准确可靠的生物样本测定方法,无法通过生物等效性研究评价受试制剂与参比制剂是否具有生物等效性。
(3)人体生物等效性研究结果显示受试制剂与参比制剂存在差异,且尚不确定该差异是否会对药物的安全有效性产生明显影响。
对于开展临床试验的情况,建议事先与监管机构沟通。
申报资料要求
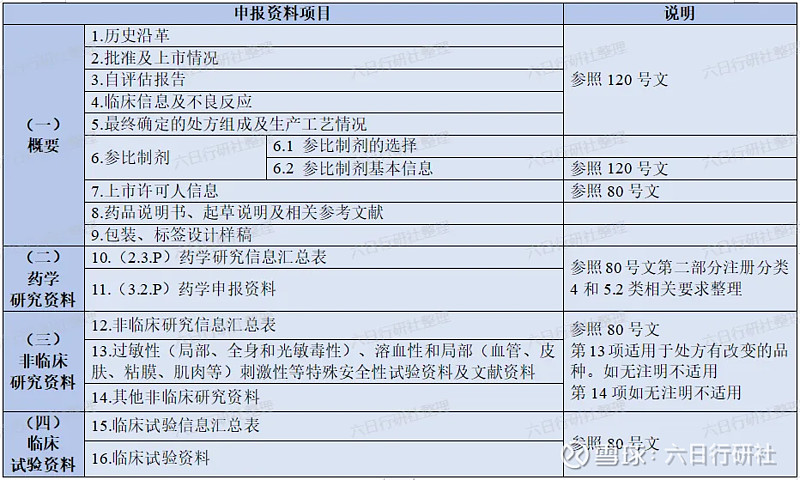
按化学药品新注册分类申报的仿制注射剂,其申报资料按照ICH M4 通用技术文件(CTD)要求整理。
一致性评价的老品种,其申报资料按照《化学药品注射剂仿制药质量和疗效一致性评价申报资料要求》整理,框架如下。
结语
以上是小编根据CDE发布的多篇指导原则进行的简单梳理,期望对化药仿制注射剂开发与一致性评价工作提供参考,如有不当之处,欢迎指正。
参考文献:
1、《化学药品注射剂仿制药质量和疗效一致性评价技术要求》
2、《化学药品注射剂(特殊注射剂)仿制药质量和疗效一致性评价技术要求》
3、《化学药品注射剂仿制药质量和疗效一致性评价申报资料要求》
4、《化学药品注射剂灭菌和无菌工艺研究及验证指导原则(试行)》
5、《化药注射剂配伍稳定性药学研究技术指导原则(试行)》
往期回顾Past Review
☀奥祺生物1200万加码生物大分子药物递送
☀网页